Not so fast, FastTree
important: see update at bottom of page Recently, much of the microbiology world has been sucked into a vortex called genomic epidemiology. This is not entirely a bad thing. Genomic data can be orders of magnitude more informative than MLST data when it comes to inferring the ancestry of an isolated microbe. Genomic data gives information on gene content. And with bacterial whole genome sequencing costs approaching parity with the cost of MLST, why sequence a few genes when you could have whole genomes? Indeed these are exciting times for microbiology. But wait, there’s a fly in the ointment.
The typical genomic epidemiology workflow goes something like this:
- sequence genomic DNA from a bunch of cultured isolates
- map each isolate’s reads to a closely related reference genome
- call variants relative to the reference
- format the variants and (hopefully!) invariant sites into a multiple alignment
- Infer a phylogeny
- Interpret the phylogeny
Much has been written about potential pitfalls in each stage of this process. For an overview of some of the issues I like the REALPHY paper. It describes the problems with leaving out invariant sites, discusses pitfalls of using a single reference genome, and even touches on the thorny issue of recombination, albeit without presenting a solution. That said, I think their software implementation leaves a bit to be desired, and Kat Holt’s blog post is worth reading for a balanced view on the single reference issue. The sky is not falling.
Last year a student I work with became involved in a genomic epidemiology project. After following a simplified genomic epidemiology workflow that carries out marker gene phylogeny based on the PhyloSift software, the student had an alignment with 21,327 columns which fall into 630 different site patterns. At my suggestion the student then applied the FastTree software to infer a phylogeny of the isolates. We used version 2.1.7. Et voilà, 55 seconds later we got a phylogeny(*):
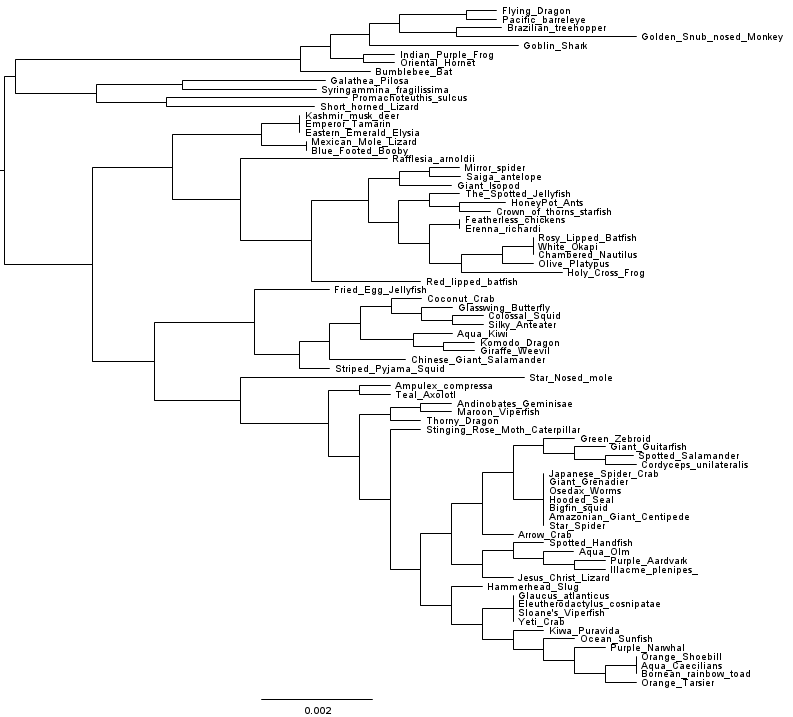
Look at that tree. Just look at it.
Does something look funny to you? The taxon names perhaps?
Look at those nice step-like internal branches in the lower half of the tree, and remember that branch lengths are given as substitutions per site – because we’re not cladists and this ain’t no freakin’ cladogram. If those branch lengths are to be believed, we have quite possibly discovered a wholly new evolutionary process. A process that regularly gives rise to divergence events after an exact number of mutations have occurred. Incredible! Stockholm is calling! Either that, or we have an inference problem. Hmmm…
Let’s dig a little deeper. What if we change the substitution model, does the tree change? What if we change the inference algorithm? We had RAxML handy so we gave that a go, and got the following tree:
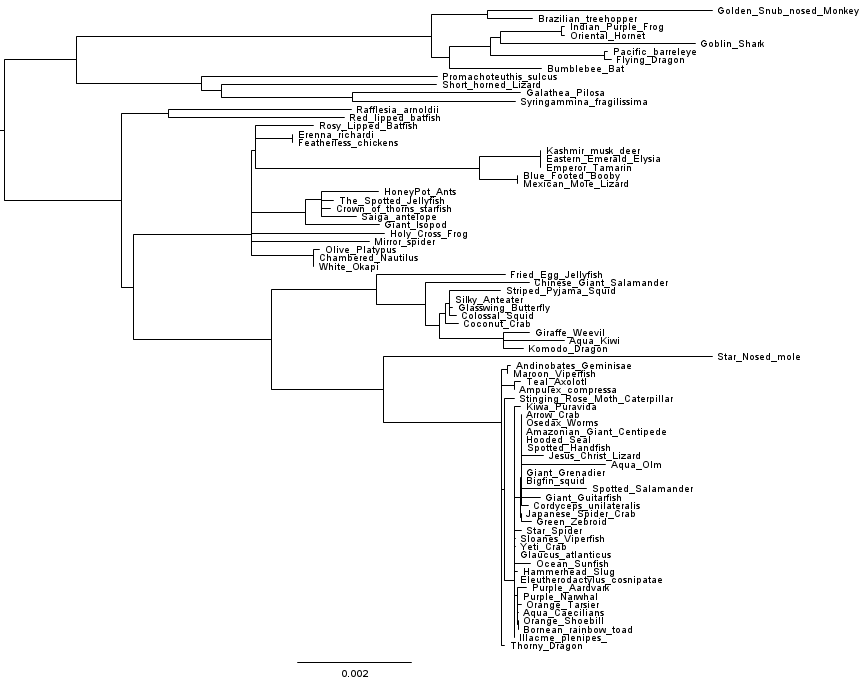
Hmmm.
What’s going on here? Which tree is right? To get some basic idea of this, Matt DeMaere (who works with me at UTS) and I had a look at how the cophenetic matrices compare to raw pairwise distance matrices derived from the genome alignments. Matt used the APE package from R to calculate cophenetic matrices from the inferred trees. Doing the comparison between the FastTree 2.1.7 cophenetic matrix, the RAxML cophenetic matrix, and the raw distance matrix, we find that the RAxML tree matches the raw distances much better than the FastTree 2.1.7 tree does.
At this stage we became concerned. After all, the venerable FastTree program is in wide use, so how could it produce such erratic results? I headed over to the FastTree web site to see if there was any documentation on the issue; any indication of a bug or a bugfix. And there, in bold header about 75% of the way down the very long page, is a section entitled Why does FastTree report so many branch lengths of 0.0005 or 0.0001, or even negative branch lengths? And there it was. The answer. It turns out FastTree by default uses single precision arithmetic and has a hard-coded limit on branch length precision. If you have data with branch lengths near that precision limit, they will either get rounded to 0 or the limit, e.g. 0.0001. This is a serious issue for many genomic epidemiology studies, where it is not uncommon to observe divergence among isolates on the order of 0.00001 to 0.0001 substitutions per site. Fortunately, the issue is easily resolved. There are a few places in the code that can be edited to enable double precision arithmetic and reduce the hard-coded limit on precision to a more reasonable value. The code diff is:
--- FastTree-2.1.7.c 2013-01-30 07:22:04.000000000 +1100
+++ FastTree_accu.c 2015-03-24 11:59:34.447129977 +1100
@@ -1,4 +1,10 @@
/*
+ * 2014/12/11 - Modifications to raise the precision of branch length
+ * estimation. This should accompany setting USE_DOUBLE.
+ * Search tolerances have been decreased to 1e-9 from
+ * their original 1e-4.
+ *
+ *
* FastTree -- inferring approximately-maximum-likelihood trees for large
* multiple sequence alignments.
*
@@ -852,10 +858,13 @@
were increased to prevent numerical problems in rare cases.
If compiled with -DUSE_DOUBLE then these minimums could be decreased.
*/
-const double MLMinBranchLengthTolerance = 1.0e-4; /* absolute tolerance for optimizing branch lengths */
+const double MLMinBranchLengthTolerance = 1.0e-9; /* absolute tolerance for optimizing branch lengths */
const double MLFTolBranchLength = 0.001; /* fractional tolerance for optimizing branch lengths */
-const double MLMinBranchLength = 5.0e-4;
-const double MLMinRelBranchLength = 2.5e-4; /* minimum of rate * length */
+const double MLMinBranchLength = 5.0e-9;
+const double MLMinRelBranchLength = 2.5e-9; /* minimum of rate * length */
+
+const double fPostTotalTolerance = 1.0e-20; /* mzd 2015/01/06, added as original assertion is violated when
+ MLMMinBranchLengthTolerance is decreased to 1e-9. */
int mlAccuracy = 1; /* Rounds of optimization of branch lengths; 1 means do 2nd round only if close */
double closeLogLkLimit = 5.0; /* If partial optimization of an NNI looks like it would decrease the log likelihood
@@ -4962,7 +4971,7 @@
double fPostTot = 0;
for (j = 0; j < 4; j++)
fPostTot += fPost[j];
- assert(fPostTot > 1e-10);
+ assert(fPostTot > fPostTotalTolerance);
double fPostInv = 1.0/fPostTot;
#if 0 /* SSE3 is slower */
vector_multiply_by(fPost, fPostInv, 4);
@@ -5025,7 +5034,7 @@
fPost[j] = value >= 0 ? value : 0;
}
double fPostTot = vector_sum(fPost, 20);
- assert(fPostTot > 1e-10);
+ assert(fPostTot > fPostTotalTolerance);
double fPostInv = 1.0/fPostTot;
vector_multiply_by(/*IN/OUT*/fPost, fPostInv, 20);
int ch = -1; /* the dominant character, if any */
These changes set the resolution limit to about 1 substitution per billion sites, which seems ample for bacteria that typically have genome sizes up to around 10 million nucleotides. It’s then necessary to compile the code with -DUSE_DOUBLE, e.g. gcc -DUSE_DOUBLE -O3 -finline-functions -funroll-loops -Wall -o FastTree-2.1.7 FastTree-2.1.7.c -lm
Let’s see how our tree looks after making these changes:
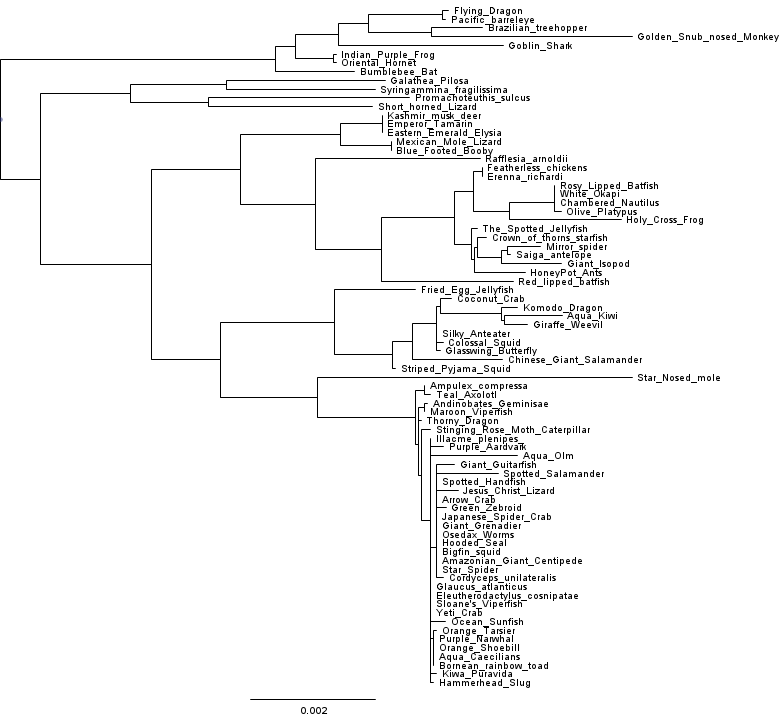
Now that looks better! And a comparison of the cophenetic matrix to the raw distances looks good too.
Another way to check whether we’ve improved model fit is to run FastTree with the -gamma option, so that the log likelihoods reported can be compared across two different runs. If we do this with the vanilla FastTree-2.1.7 it reports Gamma(20) LogLk = -40634.725. Running the same analysis with the modified double-precision FastTree yields Gamma(20) LogLk = -40182.654. In other words, the tree inferred by the double precision version is about e^452 times more likely to have generated the observed data. That’s no small improvement in model fit. Interestingly, the double-precision build actually runs faster than the single precision build on this dataset and on my laptop, so it seems the sacrifice of precision didn’t even buy us a speedup.
Perhaps this is just a case of RTFM. But how many of us actually do that? In retrospect I feel we were lucky to have identified this issue relatively early on in our data analysis. If the dataset were different, without so many genomes having divergences right around the limit of FastTree’s precision, the problem might have gone entirely unnoticed. That’s not to say it would be unimportant, though, because even just a few branches with such imprecise length estimates could in principle yield substantially reduced model fit. I am not sure to what extent this could lead to topological inaccuracy as well, but it does seem possible. Of course these issues of precision might be rather trivial compared to the effects of model misspecification, e.g. fitting a tree to a dataset with a substantial number of sites affected by recombination, and there are other tools like ClonalFrameML that implement models which address that issue. That said, even if the model is wrong (and when analyzing real data the model is wrong by definition) I’d like to have the best possible fit to the data. Wouldn’t you?
Thanks to Matt DeMaere for assistance preparing this blog post.
(*) Note that isolate names have been anonymized to protect the innocent.
Update: 25th March 2015
Morgan Price emailed to let me know that the above described changes, along with some others, have been incorporated into version 2.1.8 of FastTree. It’s available from the usual place. The increased precision is not enabled in the precompiled binaries, e.g. it’s still necessary to build with -DUSE_DOUBLE, but the precompiled binary supplies some helpful warning messages to let users know if their data may be in the danger zone:
WARNING! This alignment consists of closely-related and very-long sequences.
This version of FastTree may not report reasonable branch lengths!
Consider recompiling FastTree with -DUSE_DOUBLE.
For more information, visit
http://www.microbesonline.org/fasttree/#BranchLen
WARNING! FastTree (or other standard maximum-likelihood tools)
may not be appropriate for aligments of very closely-related sequences
like this one, as FastTree does not account for recombination or gene conversion
Happy FastTreeing!